Eliminierungsreaktion
Eliminierungsreaktion (kurz: Eliminierung) ist ein Begriff aus der organischen Chemie. Es handelt sich um einen Sammelbegriff für chemische Reaktionen, bei denen aus einem Molekül zwei Atome oder Atomgruppen abgespalten (eliminiert) werden.[1] Für diese Abspaltung müssen zwei Bindungen aufgebrochen werden. Die Eliminierung stellt die Umkehrreaktion der Addition dar.[2]
Inhaltsverzeichnis
1 Varianten
2 Reaktionsmechanismen bei der β-Eliminierung
2.1 E1
2.2 E1cb
2.3 E2
3 Konkurrenz der Reaktionsmechanismen
3.1 Optimale Bedingungen für E1
3.2 Optimale Bedingungen für E1cb
3.3 Optimale Bedingungen für E2
3.4 Stereoselektivität
4 Die nukleophile Substitution als mögliche Konkurrenzreaktion
4.1 Konkurrenz E1/SN1
4.2 Konkurrenz E2/SN2
5 Regiochemie
6 Beispiele
7 Weblinks
8 Literatur
9 Einzelnachweise
Varianten |
Die am häufigsten vorkommende Eliminierung betrifft zwei im Ausgangsmolekül benachbarte, aneinander gebundene Atome, wobei danach eine Doppel- oder Dreifachbindung zwischen diesen entsteht. Dies wird als β-Eliminierung bezeichnet.
Treten beide Abgangsgruppen an demselben Kohlenstoffatom aus, so entsteht bei dieser α-Eliminierung ein Carben.
Die γ-Eliminierung an zwei nicht direkt benachbarten Kohlenstoffatomen führt zu Cyclopropan-Derivaten.[3]
Reaktionsmechanismen bei der β-Eliminierung |
Unabhängig von den Varianten kann die Eliminierung nach verschiedenen Reaktionsmechanismen ablaufen, was in diesem Fall nicht in der Produktverteilung, sondern nur an Größen wie Reaktionsgeschwindigkeit und Aktivierungsenergie fest gemacht werden kann. Aufgrund der vorherrschenden Reaktionsbedingungen wie Substrat, angreifendes Teilchen, Temperatur und Lösungsmittel lässt sich vorhersagen, welche Eliminierung abläuft und inwiefern Konkurrenzreaktionen die Ausbeute beeinflussen.
E1 |
Der E1-Mechanismus ist eine Reaktion erster Ordnung; somit ist die Reaktionsgeschwindigkeit proportional zur Konzentration des Substrats.[4] Im Gegensatz zu anderen Mechanismen ist es für die Geschwindigkeit unerheblich, in welchen Konzentrationen das Lösungsmittel und das angreifende Nukleophil vorliegen. Eine Reaktion, die nach diesem Mechanismus verläuft, kann in zwei Teilschritte gegliedert werden.
In einem ersten, geschwindigkeitsbestimmenden Schritt wird die Abgangsgruppe abgespalten. Häufig geschieht dies, indem beispielsweise ein Halogenatom unter Aufnahme der Bindungselektronen als Halogenid-Ion austritt. Am C-Atom verbleibt daraufhin eine positive Ladung, es entsteht ein Carbokation (oder auch Carbenium-Ion genannt). Für das Kohlenstoffatom ist die Oktettregel streng gültig, sodass es bemüht ist, die Elektronenmangelsituation des Sextetts durch Aufnahme von Elektronen zu überwinden.

Im zweiten Schritt geschieht dies durch die Abspaltung eines weiteren Substituenten in β-Stellung. Dabei werden diesmal die Bindungselektronen „behalten“; die Abgangsgruppe ist positiv geladen. An der α- und β-Position findet eine Umhybridisierung des Kohlenstoffs von sp3 auf sp2 statt. Die aus der Eliminierung des Kations stammenden Bindungselektronen verteilen sich auf die beiden entstandenen, parallel zueinander stehenden p-Orbitale. Es entsteht eine C=C-Doppelbindung.[5] Oft wird im zweiten Schritt ein Proton abgespalten. Es darf nicht vergessen werden, dass dies nur formell geschieht, denn tatsächlich wird das Proton vom angreifenden Nukleophil abstrahiert. Dies ist bei einem Vergleich der Basenstärke von Halogenid-Ion und angreifendem Nukleophil schlüssig, da die stärkere Base das Proton aufnimmt.
Da intermediär ein planares Carbenium-Ion auftritt, ist durch die freie Drehbarkeit um die C-C-Achse
eine Isomerisierung möglich. Wenn keine Effekte wie sterische Hinderung ein Isomer stark destabilisieren, treten cis- und trans- Isomer mit der gleichen Wahrscheinlichkeit auf.
Es ist verständlich, dass der präparative Nutzen dieser Reaktion stark beschränkt ist, da durch die Isomerisierung nur bei stereochemisch eindeutigen Fällen ein einziges Produkt entsteht. Dies gilt auch für die nukleophile Substitution erster Ordnung. Das intermediäre Carbenium-Ion ist so reaktiv, dass neben der Problematik der Isomere ein hohes Potential für Nebenreaktionen wie Umlagerungen vorliegt.
E1cb |
Dieser Mechanismus beschreibt ebenfalls eine Reaktion erster Ordnung. Der grundlegende Unterschied zum E1-Mechanismus ist die Reihenfolge der Abspaltung. Im E1cb-Mechanismus wird zuerst das Proton und dann die Abgangsgruppe eliminiert. Es bildet sich somit ein Intermediat mit einem negativ geladenem Kohlenstoff, dem sogenannten Carbanion. Die Abkürzung cb steht für conjugated base, zu deutsch konjugierte Base, da die Reaktion über die Kohlenwasserstoff zugeordnete Base, nämlich das durch Abspaltung eines Protons entstehende Carbanion, verläuft.

Auch hier wird Stereoisomerie erst im zweiten Schritt determiniert, da eine freie Drehbarkeit um die C-C-Achse gewährleistet ist.
E2 |
Beim E2-Mechanismus verläuft die Abspaltung von Proton und Abgangsgruppe konzertiert.[4] Es gibt einen Übergangszustand, in dem das angreifende Nukleophil („Lewis-Base“) eine Verbindung mit dem Proton eingeht. Der instabile Übergangszustand wird dann über Abspaltung beider Gruppen in ein Alken überführt. Die Reaktionsgeschwindigkeit ist proportional zum Produkt aus Substratkonzentration und Konzentration der angreifenden Base.

Entscheidend für die Reaktion nach E2 ist die relative Position der beiden Abgangsgruppen. Sie müssen mit der Kernverbindungsachse auf einer Ebene liegen, also bei Aufsicht auf die C-C-Verbindungsachse in einem 0° oder 180°-Winkel zueinander stehen (siehe Newman-Projektion). Nur so können die p-Orbitale im Übergangszustand überlappen, um die Doppelbindung zu bilden.
Diese Bedingung ist bei anti- und syn-ständigen Gruppen erfüllt. Die anti-Stellung ist meist die bevorzugte, da eine schnellere Überlappung der p-Orbitale gewährleistet wird. Syn-ständige Eliminierung wird meist bei Alkanen beobachtet, deren Abgangsgruppe zugleich das Nukleophil repräsentiert.[6]
Die Stereoisomerie wird durch das verwendete Substrat oder die Eliminierungsreagenz bestimmt, da eine freie Drehbarkeit völlig ausgeschlossen ist.
Konkurrenz der Reaktionsmechanismen |
Es gibt unterschiedliche Faktoren, die Reaktion nach E1, E1cb oder E2 wahrscheinlicher machen:
Optimale Bedingungen für E1 |
Die Wahrscheinlichkeit für den Mechanismus erster Ordnung ist hoch, wenn das intermediär auftretende Carbokation stabilisiert ist:
- Ein durch Hyperkonjugation und/oder Mesomerie stabilisiertes Kation tritt auf.
- Polar-protisches Lösungsmittel: Das Kation wird durch Solvatation stabilisiert.
- Hohe Temperatur: Die Abspaltung wird wahrscheinlicher.
Schwache Basen: Starke Basen führen zu E2-Kinetik (siehe unten). Außerdem unterdrückt eine niedrige Konzentration der Base den E2-Mechanismus.
Optimale Bedingungen für E1cb |
Dieser Mechanismus konkurriert kaum mit den beiden anderen Mechanismen, da die Bedingungen zu stark voneinander abweichen. Er hat vorwiegend in der Chemie der Carbonyle eine Bedeutung:
- Die Bildung des Carbanions muss durch elektronenenziehende Substituenten stabilisiert sein. Diese machen das Auftreten eines Kations extrem unwahrscheinlich.
- Die Abgangsgruppe kann relativ schlecht sein, da durch die negative Ladung des Carbanions die Abspaltung stark begünstigt ist.
Optimale Bedingungen für E2 |
Die Mechanismen zwischen E1 und E2 ähneln sich; es kommt bei E2-Bedingungen darauf an, dass das Proton abgespalten wird, bevor sich das Carbokation ausbilden kann:
- Niedrige Temperatur und polar-aprotisches Lösemittel machen das Auftreten eines Kations unwahrscheinlich.
- Eine starke Base in hoher Konzentration greift das der elektrophilen Abgangsgruppe benachbarte Proton an, bevor es zur Dissoziation in Kation und Abgangsgruppe kommt. Ist die Base auch noch sterisch gehemmt, wird die Substitution als Konkurrenzreaktion unterdrückt (siehe unten).
Stereoselektivität |
Am Beispiel von Halogenalkanen treten die Eliminierungreaktionen in Konkurrenz.
Bevorzugung E1 vs. Bevorzugung E2 | ||||
| Halogenalkan |  | 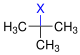 | ||
| − | primäres Halogenalkan | sekundäres Halogenalkan | tertiäres Halogenalkan | |
| Bevorzugung | E2 | E1/E2 | E1 (E2) | |
X = Halogen | ||||
Die nukleophile Substitution als mögliche Konkurrenzreaktion |
E1 steht in Konkurrenz zur SN1-Reaktion, E2 zur SN2-Reaktion. Steuern kann man dies unter anderem über Lösungsmitteleinflüsse.
Konkurrenz E1/SN1 |
E1 wird gegenüber SN1 bevorzugt, wenn ...
- Substituenten vorhanden sind, die die C=C-Bindung stabilisieren
- viele Alkylgruppen anbinden (durch den +I-Effekt)
- sperrige (sterisch anspruchsvolle) Nukleophile verwendet werden, da Bindungsaufweitung von 109,28° (sp3) auf 120° (sp2)
- schlechte Nukleophile verwendet werden (geringe Nukleophilie)
- die Temperatur erhöht wird (Entropie-Effekt)
Konkurrenz E2/SN2 |
E2 wird gegenüber SN2 bevorzugt, wenn ...
Alkyl-/Phenyl-/Vinyl-Substituenten in α- oder β-Stellung- sperrige Nukleophile verwendet werden oder bei sperrigem Kohlenwasserstoff (tertiär substituiert)
- schlechte Nukleophile anwesend
- schlechte Abgangsgruppen anwesend, da diese im Übergangszustand stärker gebunden sind
- Diederwinkel von 180/0° (anti-periplanare-Anordnung)
- stärker unpolare Lösungsmittel verwendet werden, da der Übergangszustand weniger solvatisiert ist
Bimolekulare Reaktionen sind meist eliminierungsdirigiert, da der sterische Einfluss des Übergangszustandes (SN2) sehr groß ist. Repulsive Kräfte benachteiligen einen solchen Verlauf.

Mechanismus der E2-Reaktion

Mechanismus der SN2-Reaktion
Regiochemie |
Bei sekundären und tertiären Ausgangsstoffen kann die Doppelbindung in verschiedenen Richtungen entstehen, es gibt Regioisomere. Man unterscheidet die Produkte nach der Zahl der Substituenten, die an die Doppelbindung gebunden sind als Hofmann- oder Saytzeff-Produkte (siehe auch Saytzeff-Regel nach Alexander Michailowitsch Saizew, auch Saytzeff oder Saytzev geschrieben).
Das Saytzeff-Produkt ist hierbei meist die stabilere Konfiguration, da das intermediär entstehende Carbenium-Ion, vorausgesetzt es handelt sich um eine E1-Eliminierung, durch die Substituenten stabilisiert wird (+I-Effekt). Das Hofmann-Produkt ist dann die bevorzugte Konfiguration, wenn Heteroatome in Saytzeff-Position das Carbenium-Ion destabilisieren (−I-Effekt). Des Weiteren kann es zum Hofmann-Produkt kommen, wenn – ausgehend von einer E2-Eliminierung – die Abgangsgruppe in Saytzeff-Position nicht in periplanarer Anordnung zum Proton steht, oder der Rest der Base so groß ist, dass es zu einer sterischen Hinderung kommt.
Beispiele |
| Reaktion | Abgangsgruppe | Mechanismus |
|---|---|---|
| Dehydratisierung | Wasser | E1 |
| basenkatalysierte Aldolkondensation | Hydroxidion | E1cb |
Dehydrohalogenierung zu Alkenen | Halogenid | E2 |
In der Biochemie werden α- bzw. β-Eliminierungen an Aminosäuren über Pyridoxalphosphat (Coenzym) durchgeführt.
Weblinks |
- 2D-Animation einer E1-Eliminierung
- 2D-Animation einer E2-Eliminierung
Literatur |
- Peter Sykes: Reaktionsmechanismen – eine Einführung, 8. Auflage VCH Weinheim 1982 ISBN 3-527-21090-3.
Einzelnachweise |
↑ Chemgapedia.de: Glossar:Eliminierung.
↑ A. F. Holleman, E. Wiberg, N. Wiberg: Lehrbuch der Anorganischen Chemie. 101. Auflage. de Gruyter, Berlin 1995, ISBN 3-11-012641-9, S. 384.
↑ Wissenschaft-Online-Lexika: Eliminierung im Lexikon der Chemie.
↑ ab Hans P. Latscha, Uli Kazmaier, Helmut A. Klein: Organische Chemie. 2002, Springer-Verlag, ISBN 3-540-42941-7.
↑ Chemgapedia.de: Glossar: E1-Eliminierung.
↑ Chemgapedia.de: Glossar: E2-Eliminierung.


