Katalyse

Schritte der heterogenen Katalyse
Katalyse (von altgriechisch κατάλυσις katálysis, deutsch ‚Auflösung‘)[1] bezeichnet die Änderung der Kinetik einer chemischen Reaktion mittels eines Katalysators, mit dem Ziel sie überhaupt erst in Gang zu bringen, sie zu beschleunigen oder die Selektivität in eine favorisierte Richtung zu lenken. In der lebenden Zelle spielen Enzyme, die biochemische Prozesse katalysieren, eine fundamentale Rolle im Stoffwechsel von der Verdauung bis hin zur Reproduktion und Transkription der Erbinformation. Im Umweltbereich haben sowohl natürlich ablaufende katalytische Prozesse wie die Bildung von Smog eine große Bedeutung als auch die katalytische Reduzierung von Schadstoffen im Automobil- und Kraftwerksbereich. Neue Systeme zur Energiewandlung und -speicherung wie die Brennstoffzelle basieren auf katalytischen Prozessen.
Die Wertschöpfung durch Katalyse in der chemischen Industrie ist von erheblicher volkswirtschaftlicher Bedeutung, da über 80 % aller Chemieerzeugnisse mit Hilfe katalytischer Prozesse hergestellt werden. Durch deren Optimierung kann der Energie- und Ressourcenaufwand entscheidend verringert werden. Weltweit betrug 2007 der Umsatz für Katalysatoren circa 16 Milliarden US-Dollar, wovon über 90 % mit Katalysatoren für heterogen katalysierte Prozesse erwirtschaftet wurden.
Inhaltsverzeichnis
1 Geschichte
1.1 Erste Entdeckungen
1.2 Arbeiten von Berzelius und Ostwald
1.3 Katalyse in der Nahrungsmittelherstellung
1.4 Großindustrielle Prozesse
1.5 Weitere Nobelpreise
2 Wirtschaftliche Bedeutung
2.1 Acrolein, Blausäure und Ammoniak
2.2 Stärke, Cellulose und Zucker
2.3 Fossile Rohstoffe
3 Energetische Grundlagen
4 Einteilung
4.1 Heterogene Katalyse
4.2 Homogene Katalyse
4.3 Phasentransferkatalyse
4.4 Biokatalyse
5 Katalysator
5.1 Herstellung heterogener Katalysatoren
5.2 Herstellung homogener Übergangsmetallkatalysatoren
6 Katalysatordeaktivierung und Regeneration
7 Katalytische Mechanismen
7.1 Mechanismen in der heterogenen Katalyse
7.1.1 Langmuir-Hinshelwood-Mechanismus
7.1.2 Eley-Rideal-Mechanismus
7.1.3 Mars-van-Krevelen-Mechanismus
7.2 Mechanismen in der homogenen Katalyse
7.2.1 Säure-Base-Katalyse
7.2.2 Übergangsmetallkatalyse
8 Nobelpreise im Bereich Katalyse
9 Organisationen
10 Fachzeitschriften (Auswahl)
11 Literatur
12 Weblinks
13 Einzelnachweise
Geschichte |
Erste Entdeckungen |
Als erste vom Menschen angewandte katalytische technische Prozesse gelten die Alkoholvergärung aus Zucker, von den Sumerern in Mesopotamien bereits 6000 vor Christus angewendet, sowie die Essigsäureherstellung aus Alkohol mit Hilfe von katalytisch wirkenden Enzymen. Nach diesen frühen Anfängen fand erst im 18. und frühen 19. Jahrhundert die Entdeckung einer ganzen Reihe von neuen katalytischen Reaktionen statt. So entdeckte Antoine-Augustin Parmentier 1781 die Stärkespaltung zu Zucker unter Säurekatalyse. Nur ein Jahr später entdeckte Carl Wilhelm Scheele 1782 die säurekatalysierte Veresterung von Alkoholen und Säure zu Estern und kurz darauf Joseph Priestley 1783 den Zerfall von Ethanol zu Ethylen und Wasser an Tonerde.
Als erstes Verfahren zur technischen Herstellung einer Grundchemikalie wurde von Desormes und Clement im Jahr 1806 das Bleikammerverfahren zur Herstellung von Schwefelsäure entwickelt, bei dem Stickoxide die Oxidation des Schwefeldioxids katalysieren.[2]Berthollet entdeckte den Ammoniakzerfall zu Stickstoff und Wasserstoff an Eisenkatalysatoren, 1818 Thénard den Zerfall von Wasserstoffperoxid an Silber, Silberoxid und Mangandioxid. Die von Döbereiner 1823 gefundene Entzündung von Wasserstoff an Platin führte zur Entwicklung des Döbereiners Feuerzeug, das in relativ großen Stückzahlen hergestellt wurde und bis Mitte des 19. Jahrhunderts Verwendung fand.[3]
In einem Brief an Johann Wolfgang von Goethe, seinen Dienstvorgesetzten, schrieb Döbereiner über seine Entdeckung: Gnädigster Herr Staatsminister! Ich erlaube mir, Ew. Exzellenz von einer Entdeckung Nachricht zu Geben….daß das rein metallische staubförmige Platin die höchstmerkwürdige Eigenschaft hat, das Wasserstoffgas durch bloße Berührung und ohne alle Mitwirkung äußerer Potenzen zu bestimmen, daß es sich mit Sauerstoffgas zu Wasser verbindet, wobei eine bis zum Entglühen des Platins gesteigerte Summe von Wärme erregt wird.[4]
Arbeiten von Berzelius und Ostwald |

Porträt des schwedischen Mediziners und Chemikers Jöns Jacob Berzelius, Nachdruck eines Stahlstichs
Berzelius erkannte 1835 in den obigen Reaktionen die Gemeinsamkeit, dass neben den Edukten und Produkten immer ein weiterer Stoff in der Reaktion notwendig war, der offenbar nicht verbraucht wurde. Er prägte dazu den Begriff Katalyse in Analogie zu Analyse: …Ich werde sie die katalytische Kraft der Körper, und die Zersetzung durch dieselbe Katalyse nennen. Die katalytische Kraft scheint eigentlich darin zu bestehen, dass Körper ihre blosse Gegenwart, und nicht durch ihre Verwandtschaft, die bei dieser Temperatur schlummernden Verwandtschaften zu erwecken vermögen, so dass zufolge derselben in einen zusammengesetzten Körper die Elemente sich in solche andere Verhältnisse ändern, durch welche eine größere elektrisch-chemische Neutralisierung hervorgebracht wird.[5]

Der Chemiker und Nobelpreisträger Wilhelm Ostwald auf einer Porträtfotografie
Eine moderne Definition der Katalyse fand Wilhelm Ostwald im Jahr 1894. Diese lautet: Katalyse ist die Beschleunigung eines langsam verlaufenden chemischen Vorgangs durch die Gegenwart eines fremden Stoffes.[6] Später spezifiziert zu: Ein Katalysator ist ein Stoff, der die Geschwindigkeit einer chemischen Reaktion erhöht, ohne selbst dabei verbraucht zu werden und ohne die endgültige Lage des thermodynamischen Gleichgewichts dieser Reaktion zu verändern. Als Anerkennung für seine Arbeiten über die Katalyse sowie für seine grundlegenden Untersuchungen über chemische Gleichgewichtsverhältnisse und Reaktionsgeschwindigkeiten wurde Ostwald im Jahre 1909 mit dem Nobelpreis für Chemie ausgezeichnet.
Katalyse in der Nahrungsmittelherstellung |
Neben der Weiterentwicklung der Schwefelsäureherstellung im Kontaktverfahren als großtechnisches Katalyseverfahren fand die heterogene Katalyse auch Anwendung im Bereich der Nahrungsmittelherstellung. So entdeckte Wilhelm Normann bereits 1901 die Fetthärtung durch katalytische Hydrierung von Ölsäure zu Stearinsäure mit Wasserstoff an fein verteiltem Nickel und damit die Grundlage der großindustriellen Margarineherstellung. 1909 war das Verfahren im großtechnischen Einsatz und in einer Anlage in Warrington in England wurden nach Normanns Verfahren wöchentlich 100 Tonnen Walöl zu Speisefetten verarbeitet.[7]
Schon 1903 arbeitete der französische Chemiker Victor Henri auf dem Gebiet der Enzymkatalyse. Er untersuchte die Spaltung von Saccharose mit Hilfe des Enzyms Saccharase in Glucose und Fructose. Durch die Fortsetzung seiner Arbeiten durch den deutschen Biochemiker Leonor Michaelis und die kanadische Medizinerin Maud Menten gelang 1913 die Formulierung der Michaelis-Menten-Theorie, dem bis heutige gültigen Grundstein der Enzymkinetik. Das Potential der Enzymkatalyse für die ressourcenschonende Herstellung von Feinchemikalien, Arzneimitteln, Vitaminen oder Waschmitteln ist bis heute, über 100 Jahre nach der Entdeckung der Grundlagen, bei weitem nicht ausgeschöpft.[8][9]
Großindustrielle Prozesse |
Im frühen 20. Jahrhundert begann die Entwicklung einer Reihe von Verfahren, die bis heute zu den wichtigsten der chemischen Industrie zählen. Haber, Bosch und Mittasch entwickelten 1910 die Ammoniak-Synthese aus den Elementen Stickstoff und Wasserstoff an Eisen-Kontakten, das Haber-Bosch-Verfahren. Wilhelm Ostwald entwickelte das Ostwald-Verfahren der Ammoniak-Oxidation an Platin-Netzen zu Salpetersäure, wodurch der zuvor knappe Nitrat-Dünger im großen Maßstab zur Verfügung stand.
Im Jahr 1913 wurde das erste Wacker-Verfahren zur Herstellung von Acetaldehyd aus Acetylen und Wasser an Quecksilber-Katalysatoren entdeckt. Durch das Fluid Catalytic Cracking an Silica- / Alumina-Katalysatoren wurde Benzin aus höheren Erdölfraktionen zugänglich, später durch das Hydrocracken an Nickel-Katalysatoren.[10]
Im Jahr 1923 entwickelte Matthias Pier bei der BASF ein katalytisches Hochdruckverfahren zur Synthese von Methanol aus Synthesegas an Zinkoxid-Chromoxid-Katalysatoren. Damit stand eine wichtige Grundchemikalie der industriellen Organischen Chemie zur Verfügung, die in vielen weiteren Verfahren eingesetzt wurde.[11]Fischer und Tropsch erfanden das Fischer-Tropsch-Verfahren, mit dem ab 1925 erstmals aus Kohle gewonnenes Kohlenstoffmonoxid und Wasserstoff an Eisen-Cobalt-Katalysatoren zu Paraffinen und Olefinen umgesetzt wurden. Das Verfahren gewinnt in der heutigen Zeit wieder an Bedeutung, um neben Treibstoffen auch Chemierohstoffe, etwa Olefine, aus anderen Ressourcen als Erdöl zu gewinnen. Etwa zur gleichen Zeit entdeckte Walter Reppe die homogenkatalytische Reaktion von Acetylenen mit verschiedenen Reaktanten unter Nickelkomplexkatalyse zu einem breiten Spektrum von Produkten, die so genannte Reppe-Chemie.[12]
Otto Roelen entdeckte 1938 mit der Hydroformylierung die Herstellung von Aldehyden aus Olefinen, Kohlenmonoxid und Wasserstoff an Cobalt-Katalysatoren, die er bis zum großtechnischen Prozess weiterentwickelte.[13] Die Hydroformylierung gilt als erste großtechnische Anwendung homogener Übergangsmetallkatalysatoren. Das ursprüngliche Verfahren Roelens wurde vielfach weiterentwickelt. Heute gilt das Ruhrchemie-Rhone-Poulenc Verfahren, das mit wasserlöslichen homogenen Rhodium-Katalysatoren zur leichteren Abtrennung des Katalysators arbeitet, als Stand der Hydroformylierungstechnik.
Auch auf dem Gebiet der Raffinerietechnologie wurden weitere katalytische Verfahren entwickelt. Durch Katalytisches Reforming von niedrigoktanigen Alkanen an Platin-Zinn- oder Platin-Rhenium / Aluminiumoxid-Kontakten entstanden hochoktanige, aromaten- und isoalkan-reiche Benzine. Das Verfahren stellt bis heute pro Tag mehrere Millionen Liter hochoktaniges Benzin zur Verfügung.
Mit dem von Karl Ziegler am Max-Planck-Institut für Kohlenforschung entwickelten Niederdruckverfahren, bei dem Ethylen und Propylen an Titan/Aluminium-Katalysatoren zu Polyolefinen umgesetzt werden, wurde die Grundlage für die petrochemische industrielle Massenproduktion von Polymeren gelegt, die das Kunststoffzeitalter einläutete. Ziegler wurde zusammen mit Giulio Natta für diese Arbeiten mit dem Nobelpreis für Chemie ausgezeichnet. Am MPI in Mülheim an der Ruhr entstanden auch die grundlegenden Arbeiten von Günther Wilke, der die Herstellung von 1,5-Cyclooctadien aus 1,3-Butadien an Nickel-Katalysatoren entdeckte sowie die Arbeiten von Wilhelm Keim zum SHOP-Prozess.[14]
Weitere Nobelpreise |

Asymmetrische Hydrierung nach Noyori
Das Verständnis der Enzymkatalyse und dessen Stereochemie wurde durch die Arbeiten von Cornforth, der dafür mit dem Chemie-Nobelpreis ausgezeichnet wurde, erweitert. Neben der Entwicklung der katalytischen Verfahren für Grund- und Zwischenprodukte wurden im Laufe der Jahre auch zahlreiche Verfahren für die Herstellung von Feinchemikalien entwickelt.
So wurden die Arbeiten von William S. Knowles und Ryoji Noyori „für ihre Arbeiten über chiral katalysierende Hydrierungsreaktionen“ sowie die nach Barry Sharpless benannte Epoxidierung mit dem Nobelpreis ausgezeichnet.
Ebenfalls in den 1970er-Jahren entdeckte Richard F. Heck die mit homogenen Palladium-Komplexen katalysierte Kreuzkupplung, die eine direkte Olefinierung von Arylhalogeniden erlaubt. Ein weiterer Nobelpreis auf dem Gebiet der Katalyse wurde 2005 für die Entdeckung der Alkenmetathese von Olefinen an Ruthenium-Katalysatoren an Chauvin, Schrock und Grubbs verliehen. Im Jahr 2007 wurde Gerhard Ertl für seine Studien von chemischen Verfahren auf festen Oberflächen ausgezeichnet, die weitgehende Einblicke in die Elementarschritte heterogen-katalysierter Reaktionen erlauben.[15][16]
Wirtschaftliche Bedeutung |
Die wirtschaftliche Bedeutung der Katalyse ist enorm. Die Ernährung eines großen Teils der Weltbevölkerung beruht auf mit mineralischen Düngern erzeugten Nahrungsmitteln. Der Weltmarkt für stickstoffhaltige Düngemitteln lag im Jahr 2002 bei einem Äquivalent von circa 144 Mio. t Ammoniak nach dem Haber-Bosch-Verfahren. Dafür ist etwa 1 % des globalen Energieaufwandes nötig. Das Einsparpotential durch Effizienzsteigerung der katalytischen Prozesse ist daher groß.
Im Bereich der effektiveren stofflichen und thermischen Verwertung fossiler und nachwachsender Rohstoffe sowie im Umweltbereich spielt die Katalyse eine Schlüsselrolle. Katalytische Prozesse dienen in Raffinerien zur Herstellung schwefelreduzierter und hochoktaniger Kraftstoffe. UOP, einer der führenden Hersteller von katalytischen Reforminganlagen, gibt die Kapazität der weltweit installierten CCR-Anlagen nach UOP-Patent mit über 600 Millionen Liter Benzin pro Tag an.[17]
In der Umwelttechnik trägt die Autoabgaskatalyse, die katalytische Reduktion von Stickoxiden nach dem Denox-Verfahren sowie Dieselkatalysatoren entscheidend zur Reinhaltung der Luft bei. Der Einsatz katalytischer Brenner in der Haustechnik soll durch die damit mögliche niedrigere Verbrennungstemperatur die Bildung von Stickoxiden reduzieren.
Acrolein, Blausäure und Ammoniak |
Im Bereich der Nahrungsmittelherstellung werden traditionell enzymkatalytische sowie Säure/Base-katalysierte Prozesse eingesetzt. Zunehmend werden Aminosäuren wie D,L-Methionin aus heterogenkatalytisch gewonnen Grundstoffen wie Acrolein, Blausäure und Ammoniak hergestellt. Auf dem Gebiet der Wirkstoffherstellung sind besonders Verfahren zur Herstellung enantiomerenreiner Produkte gesucht. Neben den homogenkatalytischen sind hier besonders biokatalytische Verfahren von Vorteil.
Im Bereich der nachwachsenden Rohstoffe beschränkt sich der Einsatz katalytischer Verfahren zurzeit hauptsächlich auf die basisch-katalysierte Umesterung von Triglyceriden mit Methanol zu Biodiesel. Die installierte Produktionskapazität von Biodieselanlagen betrug 2008 in Deutschland 4,85 Millionen Tonnen pro Jahr, von denen 2,8 Millionen Tonnen oder circa 8 % des Gesamtdieselbedarfs verkauft wurden.[18]
Stärke, Cellulose und Zucker |
Die nachwachsenden Rohstoffe Stärke, Cellulose und Zucker wurden überwiegend in enzymkatalytischen Fermentationsprozessen eingesetzt. Im Jahr 2007 wurden insgesamt etwa 2,1 Millionen Tonnen nachwachsende Rohstoffe stofflich in der chemischen Industrie genutzt.[19]
Fossile Rohstoffe |
Auf dem Gebiet der fossilen Rohstoffe wird intensiv nach neuen katalytischen Prozessen geforscht. Die partielle Oxidation von Methan, nach heutigen Schätzungen die mit Abstand größte ökonomisch nutzbare Kohlenwasserstoffquelle, zu Synthesegas oder die direkte heterogenkatalytische[20] oder die enzymkatalytische Oxidation mit Methan-Monooxygenase zu Methanol sind Schwerpunkte der aktuellen Forschung.
Energetische Grundlagen |

Energiediagramm einer enzymatischen Reaktion: Die Aktivierungsenergie (freie Aktivierungsenthalpie) wird im Vergleich zu unkatalysierten Reaktionen durch Stabilisierung des Übergangszustandes gesenkt. Die freie Reaktionsenthalpie dagegen bleibt unverändert.
Eine einfache chemische Reaktion A + B → AB kann zum Beispiel folgendermaßen durch einen Katalysator Kat. beeinflusst werden, wobei A-KAT dem Übergangszustand entspricht:
- A+KAT→A−KAT{displaystyle mathrm {A+KATrightarrow A-KAT!} }

- A−KAT+B→AB+KAT{displaystyle mathrm {A-KAT+Brightarrow AB+KAT!} }

Katalysatoren erhöhen die Reaktionsgeschwindigkeit von chemischen Reaktionen um mehrere Größenordnungen. Es ist jedoch nicht möglich, mit Katalysatoren Reaktionen durchzuführen, bei denen die Gesamtenergie der Endprodukte höher ist als die der Ausgangsstoffe. Solche Reaktionen sind auch mit Katalysatoren thermodynamisch verboten: Katalyse ist also eine kinetische und keine thermodynamische Erscheinung. Wie bei jeder spontan ablaufenden Reaktion muss die freie Reaktionsenthalpie (ΔG{displaystyle Delta G}

- k=A⋅e−EAR⋅T{displaystyle k=Acdot e^{frac {-E_{mathrm {A} }}{Rcdot T}}}

mit
A{displaystyle A}präexponentieller Faktor oder Frequenzfaktor, entspricht nach der Stoßtheorie dem Produkt aus der Stoßzahl Z und dem Orientierungsfaktor P,
EA{displaystyle E_{mathrm {A} }}Aktivierungsenergie (Einheit: J/mol)
R{displaystyle R}= 8,314 J/(K mol) allgemeine Gaskonstante
T{displaystyle T}absolute (thermodynamische) Temperatur (Einheit: K)
k{displaystyle k}Reaktionsgeschwindigkeitskonstante
Die Katalyse ist daher als rein kinetischer Effekt anzusehen.
Einteilung |
Die Einteilung der Katalyse erfolgt nach verschiedenen Kriterien wie den beteiligten Phasen oder der Art der eingesetzten Katalysatoren. Je nachdem, in welcher Phase Katalysator und Substrat vorliegen, wird nach homogener (Katalysator und Substrat liegen in der gleichen Phase vor) und heterogener Katalyse (Katalysator und Substrat liegen in verschiedenen Phasen vor) unterschieden. Wechselt ein Katalysator oder Substrat während der Reaktion die Phase, wird der Vorgang Phasentransferkatalyse genannt.[21]
Je nach Art des verwendeten Katalysators gibt es weitere Einteilungskriterien wie Übergangsmetallkatalyse, Säure-Base-Katalyse oder Biokatalyse. Weisen die Produkte einer katalytischen Reaktion spezielle stereochemische Eigenschaften auf, wird dies enantioselektive Katalyse genannt.[22] Wirkt ein während einer Reaktion hergestellter Stoff katalytisch auf die Erzeugungsreaktion, wird dieser Vorgang Autokatalyse genannt. Je nach Art der katalysierten Reaktion spricht man zum Beispiel von Oxidations- oder Hydrierkatalysatoren.
Werden Reaktionen mit Hilfe organischer Moleküle katalysiert, die keine Metalle enthalten, sondern nur aus leichten Hauptgruppenelementen bestehen, wird dies als Organokatalyse bezeichnet.[23] In der Organokatalyse reagiert der Katalysator mit dem Substrat zunächst unter Bildung einer kovalenten Bindung. Ein bekanntes Beispiel ist die enantioselektive Prolin-Katalyse. Darin bildet Prolin als sekundäres Amin mit einer Carbonylverbindung ein Enamin, das im tautomeren Gleichgewicht mit seinem Iminium-Ion steht. In beiden Formen kann der entstehende Komplex eine Reihe von Reaktionen eingehen, etwa nukleophile Aldol-Additionen oder Knoevenagel-Reaktionen.
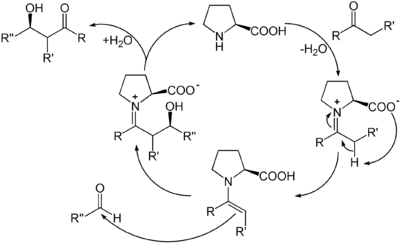
Im letzten Schritt wird durch Hydrolyse das Produkt freigesetzt und das Prolin als Katalysator zurückgewonnen.[24]
Heterogene Katalyse |
Bei der heterogenen Katalyse liegt der Katalysator oft in fester Form vor (so genannter Kontakt) und das Substrat reagiert aus der Gas- oder Flüssigphase. Die heterogene Katalyse ist die Grundlage vieler chemischer und petrochemischer Prozesse. Katalysatoren sind häufig feste Basen und Säuren, wie Zeolithe, Metalloxide oder heterogenisierte Homogenkatalysatoren, wie trägerfixierte Enzyme. Neben der Chemie der Katalysereaktion sind bei heterogen-katalysierten Reaktionen oft Transportvorgänge geschwindigkeitsbestimmende Schritte. Ein großer Vorteil von heterogenen Katalysatoren ist ihre einfache Trennung von den Reaktionsprodukten.
Die Optimierung katalytischer Prozesse erfordert einen multidisziplinären Ansatz, bei dem neben der Chemie des Katalysators und des Prozesses Aspekte der Fluiddynamik, des Reaktordesigns sowie neue Erkenntnisse der Festkörper- und Oberflächenchemie und Analytik berücksichtigt werden müssen.
Homogene Katalyse |
Bei der homogenen Katalyse können Katalysator und Substrat entweder in der Gasphase, etwa NO2 und SO2 beim Bleikammerverfahren, oder in der flüssigen Phase vorliegen. Die Selektivität ist oft höher als bei heterogen katalysierten Reaktionen, oft ist jedoch ihre Abtrennung vom entstehenden Produktgemisch schwierig.
Ist die Aktivität des eingesetzten Homogenkatalysators sehr hoch und durchläuft dieser genügend katalytische Zyklen, ist die Abtrennung des Katalysators nicht notwendig. Der Katalysator verbleibt in solchen Fällen, etwa beim Ziegler-Natta-Verfahren, in sehr geringer Menge im Produkt.
Je nach der Art des eingesetzten Katalysators werden folgende Arten der homogenen Katalyse unterschieden:[25]
- Brönsted-Säure/Base-Katalyse,
- Lewis-Säure/Base-Katalyse, oder elektrophile/nucleophile Katalyse,
- Redoxkatalyse,
- Komplexkatalyse,
- metallorganische Komplexkatalyse.
Phasentransferkatalyse |
Phasentransferkatalyse am Beispiel eines quarternären Ammoniumsalzes Q+
Vermittelt ein Katalysator den Kontakt zweier Reaktanten, die in unterschiedlichen Phasen (meist wässrige und organische Phase) vorliegen, wird dies Phasentransferkatalyse genannt. Der Katalysator einer solchen Reaktion ermöglicht den Durchtritt der Reaktanten durch die Phasengrenze.
So ermöglichen Kronenether die Lösung von Alkalimetallionen in organischen Lösungsmitteln; die Anionen, zum Beispiel MnO4-, werden als Kronenether/Alkali-Anion Ionenpaar in die organische Phase geschleppt. Dabei haben sich Kronenether mit 15 und 18 Kohlenstoffatomen im Ring besonders bewährt. Durch Einsatz chiraler Kronenether können gute Enantiomerenüberschüsse erhalten werden.[26]
Quartäre Ammoniumverbindungen, Phosphonium- oder Arsoniumionen mit lipophilen Alkylresten verbessern die Extraktion. Dabei steigt die Lipophilie mit zunehmendem Kohlenstoffanteil im Alkylrest. Die Reaktionsgeschwindigkeit einer Phasentransferkatalyse steigt oft linear mit der Konzentration des Katalysators an.
Biokatalyse |
Stoffwechselvorgänge in Lebewesen werden durch Enzyme katalysiert. Diese Reaktionen zeichnen sich allgemein durch äußerst hohe Effizienz und Selektivität aus und laufen bei milden Temperaturen und in wässrigem Milieu ab. Reaktive Spezies, die mit Wasser reagieren würden, werden durch hydrophobe „Taschen“ abgeschirmt. Viele Biokatalysatoren sind Proteine oder enthalten Proteinbestandteile. Nach ihrer Funktion werden die Enzyme in sechs Klassen als Oxidoreduktasen, Transferasen, Hydrolasen, Lyasen, Isomerasen und Ligasen eingeteilt.
Einige biokatalytische Prozesse gehören zu den ältesten bekanntesten chemischen Prozessen der Menschheit, etwa dem Bierbrauen. Biokatalytische Prozesse unter Einsatz von Bakterien, Hefen oder Pilze, besonders in der Herstellung von Wein, Bier, Käse und anderen Nahrungsmitteln sind seit langem bekannt. Auch in der Pharmazeutischen Industrie werden verstärkt biokatalytische Prozesse eingesetzt.[27] Wirtschaftlich bedeutende biokatalysierte Prozesse sind die Herstellung von Vitamin C, die Herstellung von Aspartam, die Verzuckerung von Stärke zu Glukosesirup und die Antibiotika-Herstellung durch enzymatische Spaltung von Penicillin G.
Katalysator |
Ein Katalysator ist der Stoff, der die vorbeschriebene Katalyse bewirkt. Katalysatoren finden Anwendungen für den stationären und mobilen Umweltschutz, in der chemischen Industrie und in der Raffinerietechnik. Der Umsatz mit Autoabgaskatalysatoren betrug im Jahr 2008 circa 5,2 Milliarden US-Dollar. Der Autoabgaskatalysator hatte damit den größten Anteil am Markt für heterogene Katalysatoren. Mit Katalysatoren für stationäre Umweltschutzanwendungen, etwa DENOX-Katalysatoren, wurde weltweit circa 1 Milliarde Dollar umgesetzt. Etwa 30 % des Marktes fallen auf Katalysatoren für Raffinerieprozesse wie Platforming und Catcracken, weitere 30 % werden in der chemischen Industrie in einer Vielzahl von Prozessen verwendet.
Die Effizienz beziehungsweise Aktivität eines Katalysators wird ausgedrückt durch die Wechselzahl, auch Turn Over Number genannt, beziehungsweise durch die Wechselzahl pro Zeiteinheit, die Turn Over Frequency. Eine weitere wesentliche Eigenschaft eines Katalysators ist seine Selektivität, das heißt sein Vermögen, nur die gewünschten Reaktionen zu beschleunigen, die Erzeugung unerwünschter Nebenprodukte jedoch zu vermeiden.
Oft benötigt ein katalytischer Zyklus neben dem eigentlichen Katalysator einen Cokatalysator, der die Aktivität oder Selektivität eines Katalysators in positiver Weise beeinflusst. Ein bekanntes Beispiel ist die Funktion von Methylaluminoxan als Co-Katalysator in der Olefin-Polymerisation sowie die Jodwasserstoffsäure im Monsanto-Prozess.
Herstellung heterogener Katalysatoren |
Die Wirksamkeit heterogener Katalysatoren hängt wesentlich von der Anzahl gleichzeitig möglicher Kontakte zwischen Katalysator und den Substraten ab, wofür oft eine möglichst große Katalysatoroberfläche wichtig ist. Heterogene Katalysatoren können als nicht poröse Vollkontakte oder Metallnetze wie im Ostwald-Verfahren mit geringer spezifischen Oberflächen oder als poröser Feststoff mit hoher spezifischer Oberfläche vorliegen.
Vollkontakte können beispielsweise durch Präzipitation oder Co-Präzipitation von Metallsalzen durch Alkalicarbonate oder -hydroxide hergestellt werden. Diese werden nach der Fällung gewaschen, getrocknet, calziniert und gegebenenfalls durch Reduzierung zum Metall aktiviert. Die Katalyse von Oxidationsreaktionen erfordert oft Vollkontakte, welche die aktive Komponente nur im äußeren Bereich des Katalysatorkörpers aufweisen und eine schnelle Diffusion der Reaktanten von und in den Hauptgasstrom erlauben, dem geschwindigkeitsbestimmenden Schritt bei diesen Reaktionen.
Heterogene Katalysatoren werden auf viele verschieden Arten hergestellt. Ein bekanntes Verfahren ist die Extrusion zur Herstellung von Katalysatoren für den Autoabgaskatalysator, der danach mit einem Washcoat beschichtet wird. Ein poröser Kontakt besteht entweder aus Vollmaterial wie Aluminiumoxid oder aus einem Trägermaterial, das mit einer katalytisch aktiven Komponente sowie gegebenenfalls Aktivatoren imprägniert oder beschichtet wurde.
Ein wichtiger Aspekt der porösen Kontakte besteht in der Verteilung der Porengrößen. Die Poren im Katalysatorkorn erhöhen die spezifische Oberfläche erheblich. Diese kann mehrere hundert Quadratmeter pro Gramm betragen. Nach der Definition der IUPAC unterscheiden sich drei Porengrößenbereiche: Mikroporen mit einem Durchmesser von kleiner als 2 nm, Mesoporen im Bereich von 2 bis 50 nm und Makroporen, die größer als 50 nm sind. Für das Design des Katalysators ist es wichtig, dass die Porengrößenverteilung auf die Diffusions- und Reaktionsgeschwindigkeit der Reaktion abgestimmt sind.
Nach der Imprägnierung eines Trägers mit einer Metallsalzlösung erfolgt beispielsweise die Trocknung, die Calzinierung und die Aktivierung des Metalls. Durch die spezifischen Bedingungen bei diesen Schritten lassen sich gegebenenfalls spezielle Metall-Profile auf einem Trägerpellet einstellen, die auf die Reaktionskinetik und die Diffusionscharakteristik der Reaktanten der katalysierten Reaktion abgestellt werden müssen.
Trägermaterialien tragen in der heterogenen Katalyse die feinverteilten, katalytisch wirksamen Metallcluster und können aufgrund ihrer Eigenschaften als Co-Katalysator dienen, oder sie haben als Ligand einen Einfluss auf die katalytische Aktivität des dispergierten Metalls. Bei mikroporösen Stoffen wie Zeolithen hat sich der Ionenaustausch bewährt.
Durch besondere Strukturen, zum Beispiel bei Zeolithen, können Trägermaterialien die Selektivität einer Reaktion beeinflussen. Beispiele von Trägermaterialien sind Cordierit, Ruß, Silicagel, Zeolithe oder Metalloxide wie Titandioxid und Aluminiumoxid.
Herstellung homogener Übergangsmetallkatalysatoren |
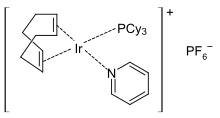
Crabtree-Hydrierungs-Katalysator
Die Herstellung homogener Übergangsmetallkatalysatoren erfolgt oft mit den Methoden der metallorganischen Chemie. Viele Übergangsmetallkatalysatoren sind luft- und feuchtigkeitsempfindlich. Eine besondere Bedeutung hat dabei das Ligandendesign für die Ausbeute und Selektivität der homogen-katalysierten Reaktion. Die elektronischen und sterischen Eigenschaften des Liganden können die Reaktion steuern und zum Beispiel sterische Informationen auf das Reaktantensystem übertragen.
Katalysatordeaktivierung und Regeneration |
Die Mechanismen der Katalysatordeaktivierung sind vielfältig. In der Übergangsmetallkatalyse wird beispielsweise die Reduktion der eingesetzten Metall-Ligand-Katalysatoren zum Metall beobachtet.
Grob kann die Art der Deaktivierung eingeteilt werden in mechanische, etwa durch Abrieb oder Zerfall, thermische wie zum Beispiel durch Sinterung, physikalische wie die Verkokung oder die physikalische Blockade aktiver Zentren sowie die chemische Deaktivierung durch Bildung inaktiver Metallkomponenten wie beispielsweise Sulfide.
In der heterogenen Katalyse sind zum Beispiel bei Raffinerieprozessen die Verkokung, die Sinterung der aktiven Oberfläche oder der Zerfall des Katalysators durch mechanischen Abrieb, zum Beispiel bei Fluid-Bed-Verfahren, bekannt. Durch Alterungsprozesse kann die katalytisch aktive Oberfläche verkleinert werden oder Poren können verstopfen, zum Beispiel in Zeolithen.
Zu den Regenerationsverfahren zählt beispielsweise das Abbrennen von Koks von Kontakten, die in Crack-Prozessen oder dem katalytisches Reforming eingesetzt werden oder die Oxychlorierung zur Wiederherstellung acider Zentren. Ist der Katalysator soweit deaktiviert, dass eine Regeneration nicht mehr sinnvoll ist, wird der Katalysator aus dem Prozess ausgeschleust. Bei Edelmetallkatalysatoren werden die Träger gegebenenfalls geschmolzen und das Edelmetall durch Verhüttungs- und elektrochemische Prozesse zurückgewonnen.
Irreversibel ist oft die Deaktivierung durch Phasenumwandlungen. Diese wird beispielsweise bei Zink-/Aluminiumoxid-Katalysatoren für die Methanolsynthese beobachtet, die zu hohen Temperaturen ausgesetzt wurden. Durch Bildung einer Spinell-Phase wird der Katalysator deaktiviert und kann nicht regeneriert werden.
Katalytische Mechanismen |
Mechanismen in der heterogenen Katalyse |
Neben den chemischen Reaktion können in der heterogenen Katalyse die Transportvorgänge die geschwindigkeitsbestimmenden Schritte sein. Es können insgesamt sieben Schritte unterschieden werden. Der erste ist die Diffusion der Reaktanten vom Gasstrom in den Katalysator. Der zweite Schritt ist die Porendiffusion, auf den die Adsorption der Reaktanten am aktiven Zentrum folgt. Nach der chemischen Reaktion verlassen die Produkte durch Desorption, wieder Porendiffusion und Diffusion in den Hauptgasstrom den Kontakt.
Der chemische Prozess nach Adsorption kann auf verschiedene Weise erfolgen. Bei monomolekularen Reaktionen zerfällt zum Beispiel das Edukt an der Katalysatoroberfläche. Bei bimolekularen Reaktionen sind drei Mechanismen denkbar, die davon abhängen, ob und wie die Reaktionspartner adsorbiert werden.
Langmuir-Hinshelwood-Mechanismus |
Der Langmuir-Hinshelwood-Mechanismus[28] besteht aus den Schritten Adsorption, Oberflächendiffusion, Oberflächenreaktion und Desorption. Zunächst müssen beide Edukte A und B aus der Gasphase auf der Katalysatoroberfläche adsorbiert werden:[29]
- Ag→Aads{displaystyle mathrm {A_{g}rightarrow A_{ads}!} }

- Bg→Bads{displaystyle mathrm {B_{g}rightarrow B_{ads}!} }

Die adsorbierten Spezies diffundieren auf der Oberfläche des Katalysators und reagieren anschließend zum Produkt C ab:
- Aads+Bads→Cads{displaystyle mathrm {A_{ads}+B_{ads}rightarrow C_{ads}!} }

Im letzten Schritt desorbiert Produkt C:
- Cads→Cg{displaystyle mathrm {C_{ads}rightarrow C_{g}!} }

Der Langmuir-Hinshelwood-Mechanismus wurde unter anderem für die Oxidation von CO an Platin-Katalysatoren, die Methanol-Synthese an Zinkoxid-Katalysatoren sowie die Oxidation von Ethylen zu Acetaldehyd an Palladium-Katalysatoren nachgewiesen.
Eley-Rideal-Mechanismus |
Beim Eley-Rideal-Mechanismus, der 1938 von D. D. Eley und Eric Rideal vorgeschlagen wurde, adsorbiert zunächst Edukt A auf der Katalysatoroberfläche:
- Ag→Aads{displaystyle mathrm {A_{g}rightarrow A_{ads}!} }
Anschließend reagiert das adsorbierte Edukt mit einem weiteren Edukt B aus der Gasphase zum Produkt C:
- Aads+Bg→Cads{displaystyle mathrm {A_{ads}+B_{g}rightarrow C_{ads}!} }

Im letzten Schritt desorbiert Produkt C:
- Cads→Cg{displaystyle mathrm {C_{ads}rightarrow C_{g}!} }
Der Eley-Rideal-Mechanismus wurde unter anderem für die Oxidation von Ammoniak an Platin-Katalysatoren sowie die selektive Hydrierung von Acetylen an Eisen-Katalysatoren nachgewiesen.
Mars-van-Krevelen-Mechanismus |
Edukt A wird zunächst aus der Gasphase auf der Katalysatoroberfläche adsorbiert:
- Ag→Aads{displaystyle mathrm {A_{g}rightarrow A_{ads}!} }
Anschließend folgt die Oxidation von Edukt A mit vorhandenem Gittersauerstoff:
- Aads+Osurf→AOads{displaystyle mathrm {A_{ads}+O_{surf}rightarrow AO_{ads}!} }

Produkt AO desorbiert und es entsteht eine Sauerstoffleerstelle im Kristallgitter:
- AOads→AOg+Leerstelle{displaystyle mathrm {AO_{ads}rightarrow AO_{g}+Leerstelle!} }

Nach der Desorption werden durch Reoxidation mit Sauerstoff die Leerstellen wieder aufgefüllt:
- O2,g→2Oads→2Osurf{displaystyle mathrm {O_{2,g}rightarrow 2;O_{ads}rightarrow 2;O_{surf}!} }

Die oxidative Dehydrierung von Propan zu Propen verläuft nach einem Mars-van-Krevelen-Mechanismus an vanadiumhaltigen Metalloxidkatalysatoren.
Mechanismen in der homogenen Katalyse |
Bekannte Formen der homogenen Katalyse sind die Säure-Base-Katalyse, die Übergangsmetallkatalyse sowie die Organokatalyse. Die Mechanismen der Organokatalyse sind vielfältig. In neuerer Zeit wurde der Einsatz chiraler Aminosäuren wie Prolin in enantioselektiven Synthesen untersucht. Gegenüber der Übergangsmetallkatalyse braucht bei der Organokatalyse kaum unter Luft- oder Wasserausschluss gearbeitet werden, die Katalysatoren sind einfach aus Naturstoffen erhältlich und in unterschiedlichen Lösungsmitteln einsetzbar.
Säure-Base-Katalyse |
Ein bekanntes Beispiel einer säurekatalysierten Reaktion ist die bereits oben erwähnte Veresterung von Carbonsäuren mit Alkoholen beziehungsweise die Umkehrung dieser Reaktion unter Verseifung. Man unterscheidet zwei Arten, die spezifische und die allgemeine Säure-beziehungsweise Basenkatalyse.
Bei der spezifischen Säurekatalyse in einem Lösungsmittel S ist die Reaktionsgeschwindigkeit proportional der Konzentration des protonierten Lösungsmittelmoleküls HS+.[30]
Die Säure selbst, AH, nimmt nur in der Gestalt an der Reaktion teil, als das sie das Gleichgewicht zwischen dem Lösungsmittel und der Säure zur protonierten Form des Lösungsmittels SH+ verschiebt:
- S+AH⟶ SH+A−{displaystyle mathrm { S+AHlongrightarrow SH^{+}A^{-}} }

In einer gepufferten wässrigen Lösung hängt die Reaktionsgeschwindigkeit des Reaktants R nur von pH-Wert des Systems ab, jedoch nicht von der Konzentration der beteiligte Säure.
- −d[R1]dt=k[SH+][R1][R2]{displaystyle mathrm {-{frac {d[R1]}{dt}}=k[SH^{+}][R1][R2]} }
![{mathrm {-{frac {d[R1]}{dt}}=k[SH^{+}][R1][R2]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/c9996175982cf896daa1b65e6634d4f550c3d000)
Diese Art der chemischen Kinetik wird beobachtet, wenn der Reaktant R1 in einem schnellen Gleichgewicht mit seiner konjugierten Säure R1H+ steht, die dann langsam mit R2 zu den Reaktionsprodukten weiterreagiert, etwa bei der Aldolkondensation.
Bei der allgemeinen Säurekatalyse tragen alle Spezies, die ein Proton zur Verfügung stellen können, zur Steigerung der Reaktionsgeschwindigkeit bei, wobei starke Säuren effektiver sind.[31]
Der Protonentransfer ist der geschwindigkeitsbestimmende Schritt in der allgemeinen Säurekatalyse, zum Beispiel bei der Azokupplung.
- −d[R1]dt=k1[SH+][R1][R2]+k2[AH1][R1][R2]+k3[AH2][R1][R2]+…{displaystyle mathrm {-{frac {d[R1]}{dt}}=k_{1}[SH^{+}][R1][R2]+k_{2}[AH^{1}][R1][R2]+k_{3}[AH^{2}][R1][R2]+dots } }
![{mathrm {-{frac {d[R1]}{dt}}=k_{1}[SH^{+}][R1][R2]+k_{2}[AH^{1}][R1][R2]+k_{3}[AH^{2}][R1][R2]+dots }}](https://wikimedia.org/api/rest_v1/media/math/render/svg/341fec73fb75a3804330d3792306c75dc430c664)
Übergangsmetallkatalyse |
Der erste Schritt der homogenen Übergangsmetallkatalyse ist die Bildung der aktiven Spezies aus dem Prä-Katalysator. Am eigentlichen Katalysator erfolgt im zweiten Schritt die oxidative Addition eines Substrats unter Änderung der Oxidationsstufe des Katalysatormetalls. In einem weiteren Schritt komplexiert ein weiteres Substrat die katalytisch aktive Spezies. Eine typische Reaktion ist im Folgeschritt die Insertion dieses Substrats in die vorher gebildete Substrat-Katalysatorbindung des oxidativ addierten Substrats. Durch Umlagerung und anschließende reduktive Eliminierung des gebildeten Produkt unter Freisetzung des ursprünglichen Katalysators wird der katalytische Zyklus geschlossen.
Beim SHOP-Prozess bildet sich die katalytisch aktive Spezies unter Abspaltung des Cyclooctadien-Liganden aus dem Nickel-Diphenylphosphinoessigsäure-COD-Komplex. Durch Insertion des Ethen in die Nickel-Wasserstoff- Bindung ein Nickel-Alkyl-Komplex, in dessen Nickel-Kohlenstoffbindung weitere Ethen-Einheiten insertieren können. Durch reduktive Eliminierung des α-Olefins wird wieder der Nickel-Wasserstoff-Diphenylphosphinoessigsäure-Komplex freigesetzt, der wieder der Ausgangspunkt für weitere katalytische Zyklen darstellt.[32]

Andere Beispiele für homogen katalysierte Prozesse sind die Hydrocyanierung, die Hydroformylierung oder die Olefinmetathese.
Nobelpreise im Bereich Katalyse |
| Jahr | Person | Begründung für die Preisvergabe |
|---|---|---|
| 1909 | Wilhelm Ostwald | „als Anerkennung für seine Arbeiten über die Katalyse sowie für seine grundlegenden Untersuchungen über chemische Gleichgewichtsverhältnisse und Reaktionsgeschwindigkeiten“ |
| 1912 | Paul Sabatier | „für seine Methode, organische Verbindungen bei Gegenwart fein verteilter Metalle zu hydrieren, wodurch der Fortschritt der organischen Chemie in den letzten Jahren in hohem Grad gefördert worden ist“ |
| 1918 | Fritz Haber | „für die Synthese von Ammoniak aus dessen Elementen“ (Haber-Bosch-Verfahren) |
| 1932 | Irving Langmuir | „für seine Entdeckungen und Forschungen im Bereich der Oberflächenchemie“ |
| 1946 | James Batcheller Sumner | „für seine Entdeckung der Kristallisierbarkeit von Enzymen“ |
| 1963 | Karl Ziegler, Giulio Natta | „für ihre Entdeckungen auf dem Gebiet der Chemie und der Technologie der Hochpolymeren“ (Ziegler-Natta-Verfahren) |
| 1975 | John W. Cornforth | „für seine Arbeiten über die Stereochemie von Enzym-Katalyse-Reaktionen“ |
| 2001 | William S. Knowles, Ryoji Noyori | „für ihre Arbeiten über chiral katalysierende Hydrierungsreaktionen“ |
Barry Sharpless | „für seine Arbeiten über chiral katalysierende Oxidationsreaktionen“ (Sharpless-Epoxidierung) | |
| 2005 | Yves Chauvin, Robert Grubbs, Richard R. Schrock | „für die Entwicklung der Metathese-Methode in der organischen Synthese“ |
| 2007 | Gerhard Ertl | „für seine Studien von chemischen Verfahren auf festen Oberflächen“ |
| 2010 | Richard F. Heck, Ei-ichi Negishi, Akira Suzuki | „für Palladium-katalysierte Kreuzkupplungen in organischer Synthese“ |
Organisationen |
Als Schnittstelle für Austausch zwischen Industrie und Hochschulen sind verschiedene Verbände im europäischen Raum auf nationaler und internationaler Ebene tätig. Die Organisationen unterstützen die Förderung von Forschung und Lehre und dienen als Interessensvertretung im Bereich Katalyse.
In Deutschland ist die Deutsche Gesellschaft für Katalyse (GECATS) tätig[33] auf europäischer Ebene vertritt die European Federation of Catalysis Societies (EFCATS)[34] wissenschaftliche Organisationen im Bereich Katalyse von 25 Ländern. Auf internationaler Ebene organisiert die International Association of Catalysis Societies (IACS), seit 1956 vor allem den International Congress on Catalysis (ICC). Nach dem vergangenen ISS-Kongress in München 2012 wird der nächste im Juli 2016 in Peking stattfinden.[35] In Nordamerika vertritt die North American Catalysis Society (NACS) die Interessen der Katalyse.[36]
Fachzeitschriften (Auswahl) |
Der aktuelle Stand der Forschung auf dem Gebiet der Katalyse wird in einer Vielzahl von Journalen und Fachzeitschriften veröffentlicht. Darunter sind die Catalysis Letters, die von Norbert Kruse und Gábor A. Somorjai herausgegeben werden und Beiträge im Bereich homogene, heterogene und enzymatische Katalyse veröffentlichen.
Die Catalysis Reviews betonen den interdisziplinären Gesichtspunkt der Katalyse und veröffentlichen Beiträge in verschiedenen Bereichen wie Reaktordesign, Computersimulation und Fortschritte in analytischen Verfahren.
Die Zeitschrift Applied Catalysis wird in einer A und B Ausgabe herausgeben. Während sich die A-Ausgabe auf den Bereich der traditionellen Katalyseforschung mit Schwerpunkt auf katalytische Zyklen im Bereich homogene, heterogene und enzymatische Katalyse sowie den Aspekten der Katalysatorherstellung, Aktivierung und Deaktivierung, Alterung und Regeneration. Die B-Ausgabe konzentriert sich auf katalytische Prozesse im Umweltbereich.
Catalysis Today veröffentlicht hauptsächlich Originalarbeiten und Reviews zu einzelnen Themen.
Das Journal of Catalysis veröffentlicht zum Beispiel Studien katalytischer Elementarprozesse an Oberflächen oder Metallkomplexen über analytische Methoden bis zu theoretischen Aspekten der Katalyse in allen Gebieten der Katalyse.
Bei Kinetics and Catalysis, einem russischen Journal, liegt der Schwerpunkt der Veröffentlichung auf dem Gebiet der kinetischen Studien und quantenmechanischen Berechnungen.
Literatur |
Katalyse allgemein
- B. Cornils, W. A. Herrmann, M. Muhler, C. Wong: Catalysis from A to Z: A Concise Encyclopedia. Verlag Wiley-VCH, 2007, ISBN 978-3-527-31438-6.
- J. Hagen: Technische Katalyse. Eine Einführung. Wiley-VCH, 1996, ISBN 3-527-28723-X, ISBN 978-3-527-28723-9.
M. Baerns: Basic Principles in Applied Catalysis. Springer, Berlin 2004, ISBN 3-540-40261-6.
Homogene Katalyse
Arno Behr: Angewandte homogene Katalyse. Wiley-VCH Verlag, 2008, ISBN 978-3-527-31666-3.
Dirk Steinborn: Grundlagen der metallorganischen Komplexkatalyse. Vieweg+Teubner, 2007, ISBN 978-3-8351-0088-6.
Heterogene Katalyse
- P. Kripylo, K.-P. Wendlandt, F. Vogt: Heterogene Katalyse in der chemischen Technik. Deutscher Verlag für Grundstoffindustrie, Leipzig 1993, ISBN 3-342-00666-8.
Gábor A. Somorjai: Introduction to Surface Chemistry and Catalysis. Wiley, New York 1994, ISBN 0-471-03192-5 (englisch).
Gerhard Ertl, Helmut Knözinger, Ferdi Schüth, Jens Weitkamp: Handbook of Heterogeneous Catalysis. Wiley-VCH, Weinheim 2008, ISBN 978-3-527-31241-2.- Gerhard Ertl: Reaktionen an Oberflächen: vom Atomaren zum Komplexen (Nobel-Vortrag). (englisch) (PDF-Datei; 666 kB)
Biokatalyse
- G. E. Jeromin, M. Bertau: Bioorganikum: Praktikum der Biokatalyse. Wiley-VCH, 2005, ISBN 3-527-31245-5.
Weblinks |
Einzelnachweise |
↑ Wilhelm Gemoll: Griechisch-Deutsches Schul- und Handwörterbuch. München/ Wien 1965.
↑ Dirk Steinborn: Grundlagen der metallorganischen Komplexkatalyse. Springer-Verlag, 2007, ISBN 978-3-8351-0088-6 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ Döbereinersche Feuerzeuge. www.gnegel.de, abgerufen am 5. Januar 2010.
↑ Briefwechsel zwischen Goethe und Johann Wolfgang Döbereiner, 1810–1830, herausgegeben von Julius Schiff, Verlag Böhlau, 1914.
↑ Rutger A. Santen, P. Van Leuwen, J. Moulijn: Catalysis. Elsevier, 2000, ISBN 978-0-444-50593-4 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ W. Ostwald: Referat zur Arbeit F. Strohmann: Über den Wärmegehalt der Bestandteile der Nahrungsmittel. In: Z. phys. Chem. 15 (1894), S. 705 f.
↑ Geschichte und Entwicklung der Fetthärtung. www.dgfett.de, abgerufen am 5. Januar 2010.
↑ Hubert Rehm: Biochemie light. Deutsch, 2008, ISBN 978-3-8171-1819-9, S. 23 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ Jan Koolman: Taschenatlas der Biochemie. Georg Thieme Verlag, 2003, ISBN 978-3-13-759403-1, S. 90 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ James E. Huheey: Anorganische Chemie. Walter de Gruyter, 2003, ISBN 978-3-11-017903-3, S. 837 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ Werner Abelshauser: Die BASF. C.H.Beck, 2002, ISBN 978-3-406-49526-7, S. 672 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ Reppe-Chemie bei der BASF@1@2Vorlage:Toter Link/www.basf.com (Seite nicht mehr abrufbar, Suche in Webarchiven) Info: Der Link wurde automatisch als defekt markiert. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis.
Info: Der Link wurde automatisch als defekt markiert. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis.
↑ Boy Cornils, Wolfgang A. Herrmann, Manfred Rasch: Otto Roelen als Wegbereiter der industriellen homogenen Katalyse. In: Angewandte Chemie. 106, 1994, S. 2219–2238.
↑ Boy Cornils: Aqueous-Phase Organometallic Catalysis. John Wiley & Sons, 2004, ISBN 978-3-527-30712-8, S. 639 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ Thomas James Lindberg: Strategies and Tactics in Organic Synthesis. Academic Press, 2004, ISBN 978-0-12-450283-3, S. 316 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ Informationen der Nobelstiftung zur Preisverleihung 2007 an Gerhard Ertl (englisch)
↑ CCR Platforming. www.uop.com, archiviert vom Original am 9. November 2006; abgerufen am 31. Dezember 2009 (PDF). Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis.@1@2Vorlage:Webachiv/IABot/www.uop.com
↑ renewables Made in Germany: Biokraftstoffe Allgemein. www.renewables-made-in-germany.com, archiviert vom Original am 24. Februar 2015; abgerufen am 5. Januar 2010. Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis.@1@2Vorlage:Webachiv/IABot/www.renewables-made-in-germany.com
↑ Nachwachsende Rohstoffe in der Industrie. www.fnr-server.de, abgerufen am 5. Januar 2010 (PDF; 4,1 MB).
↑ Max-Planck-Gesellschaft – Ein Pulver gegen Energieverschwendung. www.mpg.de, abgerufen am 5. Januar 2010.
↑ C.M. Starks: Phase-Transfer Catalysis. Springer Science & Business Media, 1994, ISBN 978-0-412-04071-9 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ Ryoji Noyori: Asymmetric Catalysis: Science and Opportunities, Nobel-Vortrag, 8. Dezember 2001. nobelprize.org, abgerufen am 21. November 2009 (PDF; 879 kB).
↑ Skript der Universität Hannover über Organokatalyse (Memento vom 11. Juni 2007 im Internet Archive)
↑ U. Eder, G. Sauer, R. Wiechert: New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. In: Angew. Chem., Int. Ed. 1971, 10, S. 496
↑ Dirk Steinborn: Grundlagen der metallorganischen Komplexkatalyse. Verlag B. G. Teubner, 2007, ISBN 978-3-8351-0088-6, S. 8.
↑ PTCIssue17.pdf (application/pdf-Objekt). www.phasetransfer.com, abgerufen am 4. Januar 2010 (PDF; 351 kB).
↑ Manfred T. Reetz: Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future. In: J. Am. Chem. Soc. 135 (2013) S. 12480–12496.
↑ Eintrag zu Langmuir–Hinshelwood mechanism. In: IUPAC Compendium of Chemical Terminology (the “Gold Book”). doi:10.1351/goldbook.L03451 .
↑ Kurt W. Kolasinski: Surface Science. John Wiley & Sons, 2008, ISBN 978-0-470-99781-9, S. 183 (eingeschränkte Vorschau in der Google-Buchsuche).
↑ Eintrag zu Specific Catalysis. In: IUPAC Compendium of Chemical Terminology (the “Gold Book”). doi:10.1351/goldbook.S05796 Version: 2.3.1.
↑ Eintrag zu General Acid Catalysis. In: IUPAC Compendium of Chemical Terminology (the “Gold Book”). doi:10.1351/goldbook.G02609 Version: 2.3.1.
↑ W. Keim, F. H. Kowaldt, R. Goddard, C. Krüger: Neuartige Koordinierungsweise von (Benzoylmethylen)-triphenylphosphoran in einem Nickel-Oligomerisierungskatalysator. In: Angewandte Chememie. 1978, 90, S. 493.
↑ ,
GECATS – Deutsche Gesellschaft für Katalyse – German Catalysis Society. www.gecats.de, abgerufen am 26. Dezember 2009.
↑ EFCATS – European Federation of Catalysis Societies. www.efcats.org, abgerufen am 26. Dezember 2009.
↑ IACS – the International Association of the Catalysis Societies. www.iacs-icc.org, abgerufen am 5. Januar 2010.
↑ The North American Catalysis Society. www.nacatsoc.org, abgerufen am 5. Januar 2010.


